Sickle cell disease (SCD) is a genetic disease caused by mutations in the HBB gene, which gives instructions for making beta-globin, one of the subunits of hemoglobin. Hemoglobin is a protein in red blood cells and is in charge of transporting oxygen. It’s made up of 4 subunits, and beta-globin makes up 2 of these subunits. When the HBB gene gets mutated, the normal beta-globin subunit turns into hemoglobin S (HbS). When HbS is the version of beta-globin in hemoglobin, red blood cells from their normal round shape into a crescent, sickle-like shape. These sickle cells tend to break down prematurely, resulting in a low red blood cell count, or anemia. Anemia can cause things like shortness of breath, fatigue, dizziness, and other symptoms. Sometimes, the sickle-shaped cells can get stuck in small blood vessels and cause lots of pain (sickle cell crisis). This also stops red blood cells from reaching organs and tissues, cutting them off from oxygen and causing damage. In very serious cases, these blockages can cause high blood pressure in the blood vessels supplying blood to the lungs, leading to heart failure. As blockages can sometimes happen in the brain, people with SCD are also at a greater risk for stroke. SCD is most common in people with African ancestry, and according to an estimate by the Sickle Cell Disease Association of Canada, there are currently around 5000 people with SCD in Canada.
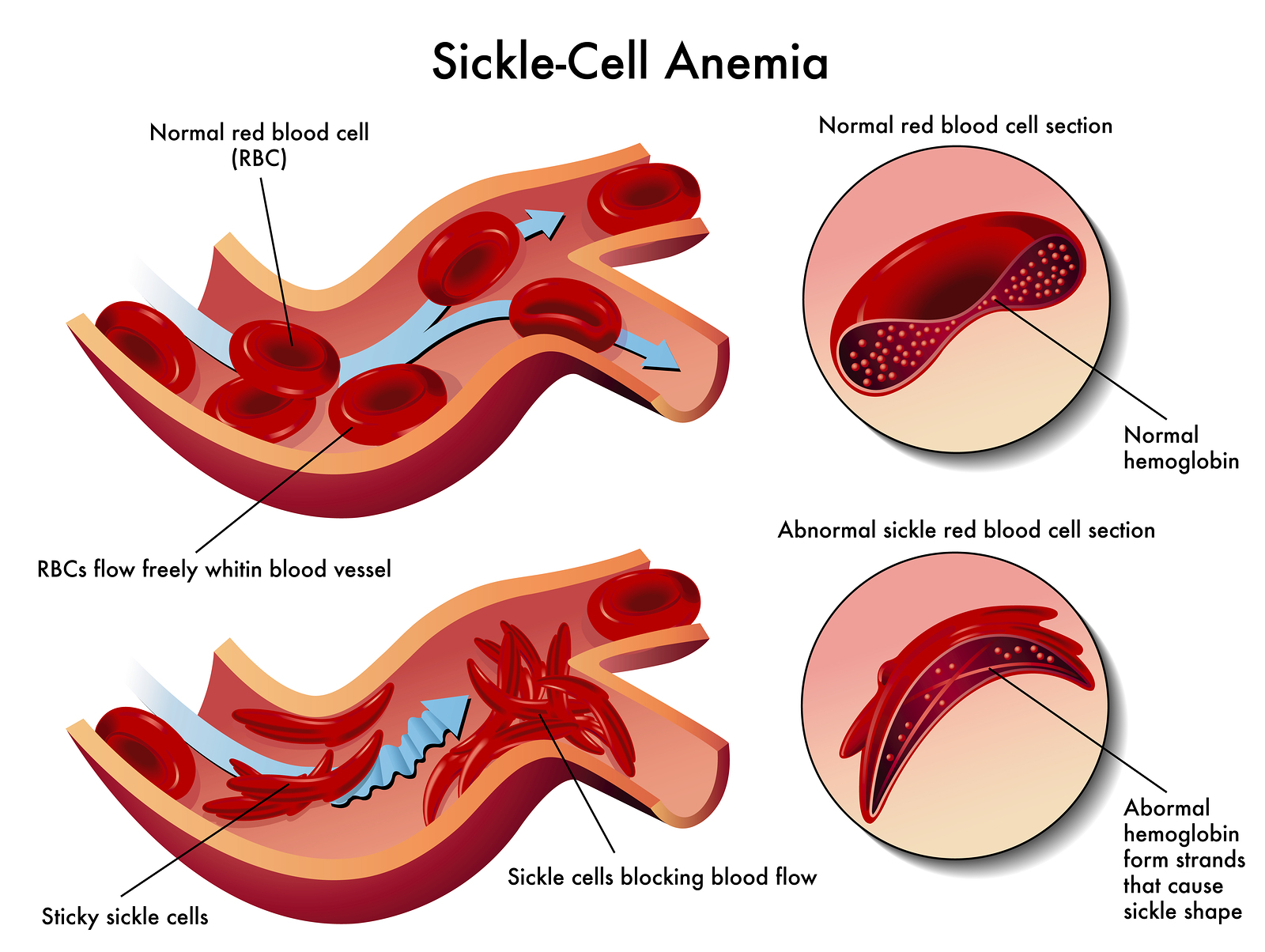
Difference between sickle cell and normal blood cells By Diana grib [CC BY-SA 4.0 (https://creativecommons.org/licenses/by-sa/4.0)], from Wikimedia Commons
As noted before, stem cell transplant is currently the only way to completely cure SCD. The procedure is usually patients with severe cases of SCD. First, chemotherapy is used to kill the bone marrow that make red blood cells with mutated hemoglobin. Then, healthy bone marrow cells taken from a donor are transplanted to the patient, usually through an IV. The donor is usually a family member of the patient. The entire transplant process starting from chemotherapy treatment to when it is complete can take months. There is also the risk of the patient rejecting the donor’s bone marrow, so recipients have to take immunosuppressant drugs for years afterward. This original treatment involving chemotherapy was often considered too dangerous for adults, who are more weaken by SCD than children and also at a higher risk for rejection.
In 2014, researchers at the NIH Clinical Center in Bethesda, Maryland released results of a clinical trial of a new stem cell transplant plan that didn’t involve chemotherapy and could be used for adults. This new plan still involved using immunosuppressants and a low dose of radiation to kill off some of the patient’s bone marrow cells, but was less devastating on the body compared to chemotherapy. The clinical trial took place from 2004 to 2013, and a total of 30 adult patients were treated using this method. 26 of the patients were successfully cured of SCD. The patients didn’t have any rejection in follow ups 3 years after the treatment, and 15 of them were able to stop taking immunosuppressants after a year. This method was not only able to cure adults, but was also safer for children.
Back in 2009, Alberta Children’s Hospital began treating children and teenagers with SCD using this new method. As of 2016, this treatment has managed to cure 9 people of SCD. But until 2018, this procedure has never been used to treat an adult in Canada. All of this changed in April 3, when Revée Agyepong became the first adult in Canada to be cured of SCD. Revée is 26 years old and had been told before that she was too old to receive transplant treatment because of the increased risks. However, with this new treatment plan, Revée was able to undergo a transplant at the Tom Baker Cancer Centre in Calgary using bone marrow cells donated by her sister. On April 3, her blood tests revealed that amount of sickle cells in her blood has decreased almost to zero, and she was essentially cured of SCD.
SCD can be a devastating disease, dramatically decreasing quality of life and causing serious complications for those who have it. But with these new treatments, the lives of people suffering from SCD can be significantly improved.

Recent Comments