
Introduction
With 45% of the composition of human blood being different types of blood cells, it’s pretty astonishing to learn that nearly all of those cells are red blood cells (more than 99%!). Having this knowledge, it seems safe to assume that they must be important molecules. These non-nucleated discs are responsible for the carrying of oxygen and carbon dioxide throughout the human body. They are able to do this because of a protein called hemoglobin, with a complex structure that we seem to take for granted. But what happens if this structure ends up being made wrong, or isn’t made at all? There are many variations of hemoglobin that can arise, and unfortunately, if you are born with one, it means you are born with a hemoglobinopathy.
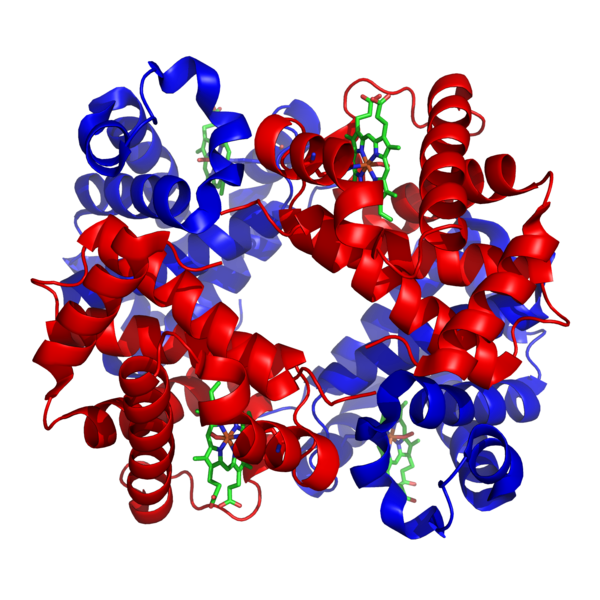
Figure 1. 1GZX Haemoglobin.png by Richard Wheeler (Zephyris). CC BY-SA 3.0 (https://commons.wikimedia.org/wiki/File:1GZX_Haemoglobin.png).
What Are Hemoglobinopathies?
Hemoglobinopathies are genetically inherited, autosomal recessive disorders. There are two main categories: Sickle cell disease, and thalassemias. According to the World Health Organization, 5% of the world’s population carries the trait genes for them, though the diseases are most prominent in those of African, Mediterranean, South Asian, and Middle Eastern descent. Sickle cell anemia is also common in people from places such as South America, the Caribbean, and Central America.
What Causes Them?
Sickle cell anemia is caused by a single nucleotide mutation in the hemoglobin beta gene on chromosome 11, position 6. This results in a valine being made instead of a glutamic acid. Though the change is small, it has big consequences for hemoglobin, causing the red blood cells to take on a rigid, crescent shape and making travel through blood vessels difficult. A little more goes into the cause of thalassemia. There are two variations of this disorder, known as alpha and beta. The type a person inherits is dependent on which part of the hemoglobin protein is being formed defectively: the alpha or beta globin protein chains. The alpha globin protein chains require four genes, and the beta globin protein chains require two. The number of genes missing determines the severity of the illness. The result of having this disease is less hemoglobin, and fewer healthy erythrocytes.
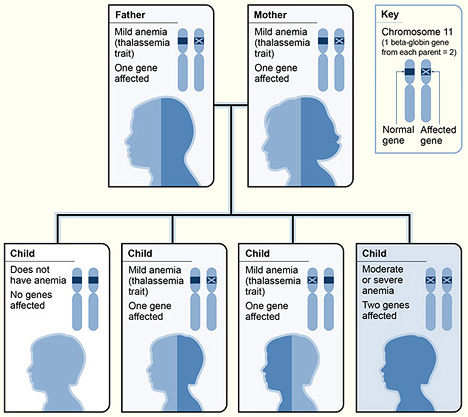
Figure 2. Thalassemia_beta.jpg by the National Heart Lung and Blood Institute (NIH). Public domain (https://commons.wikimedia.org/wiki/File:Thalassemia_beta.jpg).
What Are The Symptoms?
Though some symptoms and complications, such as anemia and increased risk of infections are shared by the two categories, some complications are more specific. For example, those with sickle cell disease are prone to painful episodes called crises when the malformed red blood cells block blood flow in the vessels and reduce oxygen delivery. The blockage can also cause strokes. Those living with thalassemia may develop bone problems like osteoporosis. This is due to the bone marrow having to work harder to make blood cells. The excess work causes the marrow to grow larger, resulting in thinner and weaker bones. Other organs affected by hemoglobinopathies include the spleen, heart, and liver, and that’s not even all of them.
What Can Be Done About Them?
Thankfully these diseases can be diagnosed early in life, at birth for sickle cell disease, and in the early childhood years for thalassemia. However, if you are concerned about your child possibly having one of these diseases, prenatal testing is available. Early diagnoses allow treatments to begin as soon as possible. Many options are available, but research is still being done in the field of hemoglobinopathies. At the moment, the only known curative treatment is hematopoietic stem cell transplantation, which can have complications, including increased risk of developing oral or esophageal cancer. And while there are many medications that can be prescribed to help with dealing with symptoms and complications, only one has officially been approved by the FDA for sickle cell anemia. Known as hydroxyurea, it was more commonly used for adults. However, a recent study has found it to be just as effective at reducing chances of stroke in children as blood transfusions. Studies are being performed with a new drug that was recently developed called GBT400, which has been found to modify hemoglobin in such a way that oxygen binds tighter to the protein. They hope it can be used to directly treat the disease, not just the symptoms. Gene therapies are also being attempted, but this line of research is getting more satisfying results for thalassemia treatment.
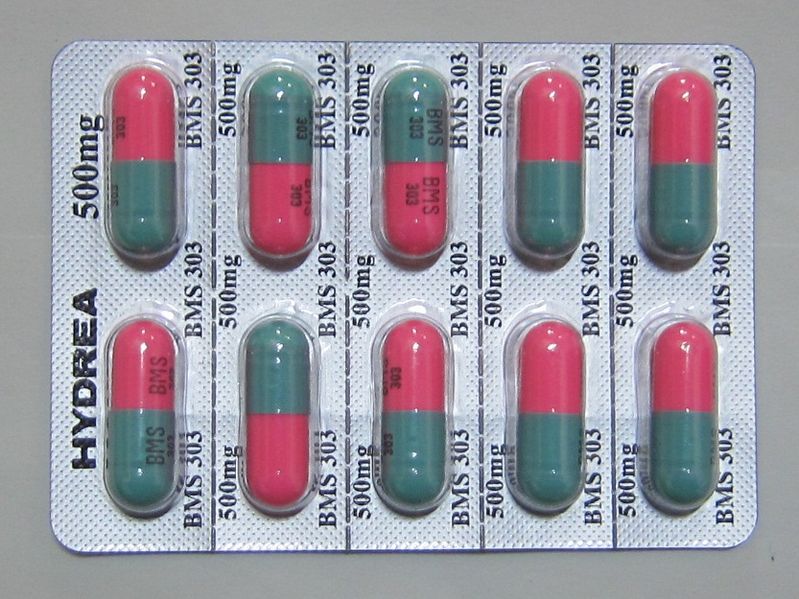
Figure 3. HYDREA (ハイドレア).jpg (HYDREA 500mg capsule (Packing for Japan)) by ぱたごん (Patagon). CC BY-SA 3.0 (https://commons.wikimedia.org/wiki/File:HYDREA_(%E3%83%8F%E3%82%A4%E3%83%89%E3%83%AC%E3%82%A2).jpg#filehistory).
Conclusion
So the next time you find yourself thinking about red blood cells and hemoglobin, don’t just think of the fact that they’re the reason our blood is red. Think of the complex structure involved, and how easily its formation can go wrong and cause problems for our bodies.
Recent Comments