The world showcases a seemingly endless number of disorders and diseases that plague humanity, from the presence of multiple limbs, to reptile like skin shedding in infants (lamellar ichthyosis), in my opinion none are more intriguing than fibrodysplasia ossificans progressive (FOP). The earliest documented cases of FOP date back to the 17th century with one such practitioner John Freke (1736) describing the disease in a 14 year old boy, “They arise from all the Vertebrae of the Neck, and reach down to the Os Sacrum; they likewise arise from every Rib of his Body, and joining together in all Parts of his Back, as the Ramifications of Coral do, they make as it were, a fixed bony Pair of Bodice.” FOP also known as stone man syndrome, baffled the medicinal community and until recently was regarded as one of medicines most elusive mysteries.
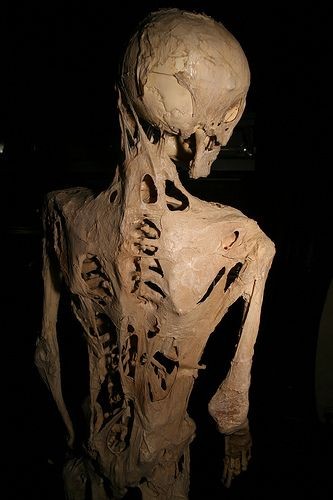
The effects of fibrodysplasia ossificans progressiva. Accessed February 25, 2016.
https://commons.wikimedia.org/wiki/File:Fibrodysplasia_ossificans_progressiva.jpg
It is difficult to imagine becoming imprisoned within our own skeleton, the very structure that facilitates mobility and our freedom to do as we please; but for those suffering from fibrodysplasia ossificans progressiva (FOP) this is their reality. FOP is a rare autosomal dominant disease that results in progressive heterotopic ossification, or more simply put, irregular bone formation. A mutation in the body’s skeletal repair mechanism causes skeletal muscle and soft connective tissue such as fascia, ligaments, and tendons to be gradually encased in bone (ossified). The ossification events are sporadic occurring primarily as a result of minor injury including muscle trauma or fatigue, even bruises and intramuscular immunization can result in ossification. Each ossification event reduces the individual’s mobility, progressing through life until the only achievable movement is in the lips and eyes. The process of ossification does not affect smooth muscle or cardiac muscle and interestingly doesn’t seem to affect tissue in the diaphragm, tongue, or extra ocular muscle.
FOP is a rare disease with approximately 1 case per two million people. FOP displays no ethical, gender, racial, or geographic predisposition, with new cases generally arising through spontaneous mutation in offspring. The two primary clinical features presented by the disease are the disfigurement of the great toes, and heterotopic ossification in characteristic patterns, beginning in the neck and upper back and progressing into more distal tissues. The malformation of the big toes upon birth is characteristic in all diseased individuals, but is often overlooked in diagnosis. At a young age FOP individuals develop swellings in soft tissues that are often misdiagnosed as tumors, subsequent biopsies and surgery lead to the explosive progression of the disease resulting in rapid bone growth. The disease inevitably progresses through life, ossifying muscle groups gradually and typically confining the individual to a wheelchair by the age of 30, and resulting in death by the age of 45. Fatalities primarily result due to thoracic insufficiency syndrome, were ossification of the rib and back muscles inhibit normal breathing.
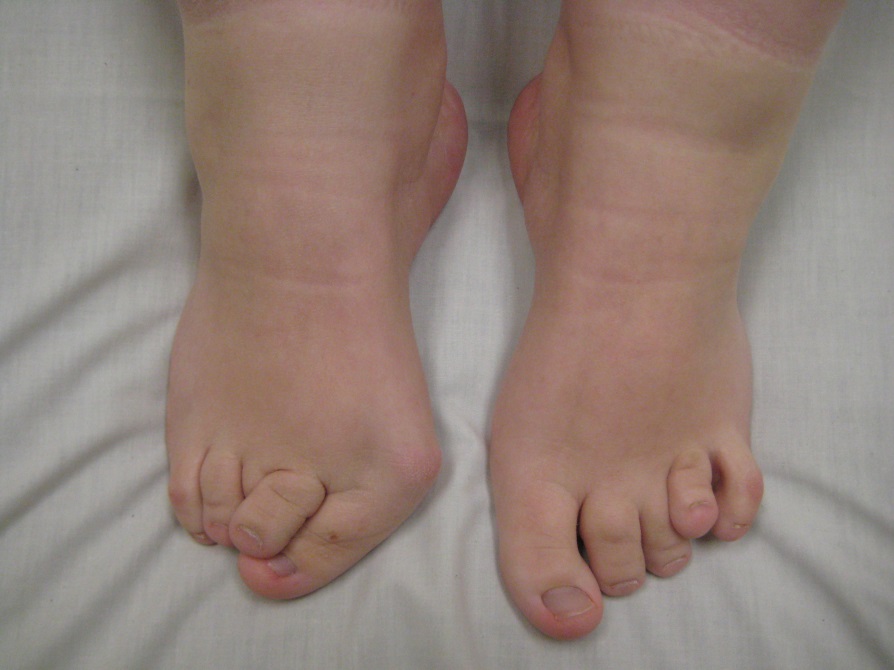
Characteristic malformation of big toe upon birth. Accessed February 26,2016
https://en.wikipedia.org/wiki/Larsen_syndrome#/media/File:Feet_of_a_person_with_Larsen_syndrome.jpg
The heterotopic ossification seen in FOP results due to the over activation of the bone morphogenetic protein (BMP) signaling pathway. BMP’s are a group of growth factors attributed with a wide variety of cellular functions including the ability to induce bone formation. BMP’s are produced by osteoclasts and perform several essential operations in embryonic development including the development of the heart, CNS, cartilage, and post-natal bone development. In order to activate the BMP signaling pathway associated with development, BMP’s must bind the BMP receptor present in cell membranes; once activated BMP receptors phosphorylate intracellular SMAD proteins among others, activating the BMP signal cascade. BMP concentrations decrease after the completion of embryonic and post-natal development in humans inhibiting certain forms of growth and development.

3D-reconstructed CT scan showing bilateral ligament ossification. Accessed February 26, 2016
https://en.wikipedia.org/wiki/Eagle_syndrome#/media/File:Verknoechertes_Lig_stylohyoideum_-_CT_3D.jpg
Analyses of individuals with FOP have identified a recurrent mutation in the 2q23-24 genetic region encoding the BMP type 1 receptor known as IA/activin-like kinase 2 (ACVR1). ACVR1 is a BMP receptor able to phosphorylate SMAD proteins 1-5 upon activation. The mutation in the FOP gene induces an alteration in the amino acid sequence, switching an arginine residue to a histidine residue at the 206 position. The alteration destabilizes the GS domain of ACVR1 responsible for activation of the SMAD proteins, subsequently causing the continuous activation of SMAD proteins and therefor the BMP signaling pathway. The R206H substitution represents a gain of function mutation allowing the ACVR1 receptor to activate SMAD proteins 1-5 and corresponding osteogenic activity without the presence of BMP.
There is currently no standardized treatment for FOP; treatment generally focuses on managing the diseases progression by limiting ossification provoking events. The recent discovery of ACVR1 over activation as the causal for FOP has made this receptor the primary therapeutic target. The latest research regarding FOP treatment has demonstrated the protein activin A acts as a secondary substrate for mutant but not wild type ACVR1 activation. The team utilized anti-activin A antibodies in conjunction with BMP blockers in mice with FOP to decrease activin A concentrations, resulting in decreased heterotopic ossification within the mice. Through recent advancements in our knowledge concerning FOP and the ongoing research towards effective treatments, individuals suffering from this devastating disease have an opportunity for hope. Research and experimentation have opened the door to this once mysterious disease, allowing science to gain a foothold, and make strides towards alleviating those suffering from FOP.
Recent Comments